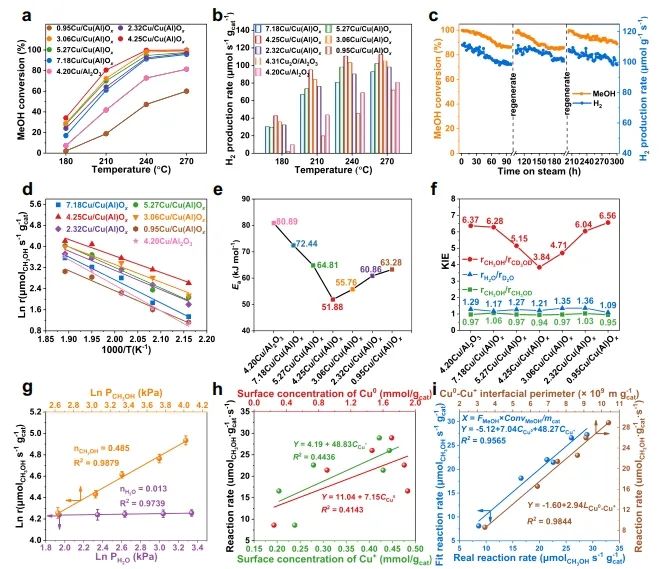
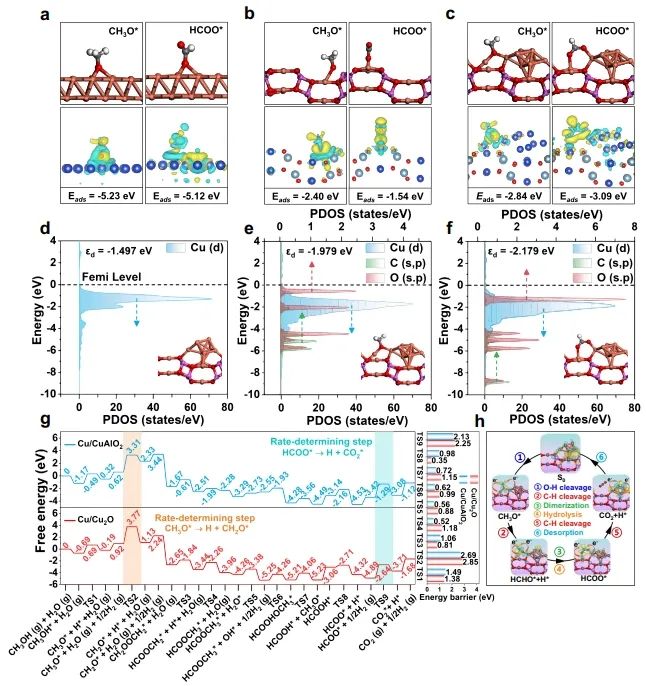
1. Nat. Commun.: 構建Cu0-Cu+雙位點,有效改善MSR反應中C-H鍵斷裂 隨著資源和環境問題的日益嚴峻,氫能被認為是化石能源的有效替代品。其中,甲醇(CH3OH)水蒸氣重整(MSR)由于能耗較低和操作簡單而成為一種經濟有效的產氫方法。在催化MSR反應的各種催化劑中,Cu具有成本效益、良好的低溫活性和高H2選擇性等優點。由于Cu具有豐富的氧化還原性質,在實際反應條件下,多種銅物種(Cu0,Cuδ+/Cu+)通常共存;此外,合金中的電子重排效應、強烈的金屬-載體相互作用(SMSI)和氧空位誘導使得揭示本征活性位點、吸附行為和反應機理等基礎問題相當復雜,導致催化性能與微觀結構之間的關系不明確。因此,揭示催化劑的本征活性位點、構效關系和反應機理不僅可以為多相催化劑的結構設計提供合理的依據,而且可以促進MSR實際應用的進一步發展。近日,北京化工大學衛敏、張建、楊宇森和浙江大學肖豐收等通過共沉淀法和后續還原處理制備了一系列具有可調Cu0-Cu+位點的yCu/Cu(Al)Ox樣品(y表示Cu/Al的質量比),并通過原位表征和理論計算研究了催化劑的活性中心。實驗結果表明,在240 ℃下,4.25Cu/Cu(Al)Ox催化劑表現出最佳的MSR催化性能,CH3OH轉化率>99%,H2產率高達110.8 μmol s-1 gcat-1,這優于先前報道的用于MSR的Cu基催化劑。此外,4.25Cu/Cu(Al)Ox催化劑的CH3OH轉化率和H2產率在100 h后有所下降(分別從99.5%和110.8 μmol s-1 gcat-1降至86.3%和99.4 μmol s-1 gcat-1),但經過再生(300 ℃空氣氧化1 h,220 ℃下25% H2/N2還原1 h)后,催化性能可恢復到原來的水平。動力學研究、原位FT-IR光譜和質譜法分析證實,4.25Cu/Cu(Al)Ox催化劑上的MSR反應經歷三個主要過程:CH3OH脫氫、HCOOCH3水解和HCOO*分解,其中CH3O*和HCOO*中間體的C-H鍵斷裂是速率控制步驟。值得注意的是,Cu0-Cu+界面協同催化起決定性作用:含氧中間體(CH3O*和HCOO*)在Cu0-Cu+界面位置發生吸附活化,適中的吸附強度引起催化劑界面的重構以及從催化劑界面到反應中間體的電子轉移;幾何結構和電子結構的變化導致C-H鍵斷裂的能壘降低,顯著促進了MSR反應。綜上,該項工作在原子水平上揭示了MSR中Cu0-Cu+界面的協同效應,這可能為合理設計高性能的MSR催化劑提供指導。Designing Cu0?Cu+ dual sites for improved C?H bond fracture towards methanol steam reforming. Nature Communications, 2023. DOI: 10.1038/s41467-023-43679-0 2. Nat. Commun.: 理論計算結合實驗,揭示非氧化還原Ni3+催化有機物的親核電氧化大多數有機物電氧化所需的電位遠低于高能耗的水氧化。因此,用熱力學上有利的有機物電氧化取代水分解OER提供了一種節能的制氫策略,并同時能夠生產高附加值化學品或處理工業廢水。Ni基電催化劑廣泛應用于大規模的工業水電解,并且在有機物的電氧化中也具有高活性。然而,在Ni基催化劑上的水氧化和有機物氧化電化學過程有很大的不同:對于Ni(OH)2電極,水氧化通常發生在Ni2+(OH)2/Ni3+OOH氧化之后,并且它們的電位差足夠大以至于可以使用Ni(OH)2/NiOOH作為氧化還原對解耦OER和析氫反應(HER)。這意味著,盡管Ni2+(3d8,t2g6eg2)通過Ni(OH)2→NiOOH+h++e?氧化為Ni3+(3d7,t2g6eg1),但是NiOOH仍然不能像高價Niδ+(δ>3)一樣氧化水。相反,與OER過程完全不同,一旦Ni(OH)2被氧化成NiOOH,有機物立即被電解。然而,由于在Ni基催化劑上水和有機物氧化的性質存在較大差異,催化機理尚不清楚,極大地阻礙了高效催化劑的設計和制備。最近,南京大學閆世成課題組使用各種理論和實驗技術,清楚地表明有機物在NiOOH上的電氧化不遵循Ni(OH)2/NiOOH氧化還原介導的電子轉移機制,而是沒有Ni3+物種氧化狀態變化的非氧化還原電子轉移過程。Ni3+作為親電電氧化中心,與具有最高占據分子軌道(HOMO)能級(?7.4到?6 eV)的有機物(親核官能團中親核原子的雙局部軟度值(?sk)為?0.65到?0.15)形成等能轉移通道。因此,(HOMO,?sk)組合判據可以很好地解釋為什么Ni3+不能有效地氧化水,但對有機物的電氧化起作用。有機物的快速電氧化動力學可歸因于親核攻擊Ni活性位建立能量轉移通道,以及有機物HOMO能級電子的化學勢足夠高,可以驅動電子從成鍵軌道向催化中心轉移。雖然水中O原子的?sk為?0.607,但水的HOMO能級(?7.99 eV)低于NiOOH的費米能級,觸發水氧化電子轉移的熱力學要求較高。因此,可以清晰地描述完整的電化學電子轉移過程:對于電催化氧化反應來說,外加電壓首先將活性中心從低價態升高到高價態,此過程可能伴隨質子耦合電子轉移的相變過程;高價態催化活性中心與反應中間體通過軌道交疊形成成鍵軌道作為電子轉移的能量通道,二者之間電子轉移遵循Marcus電荷轉移理論,而催化中心則通過高價態活性離子的未占據軌道直接轉移電子,此時催化中心并不發生價態變化,通常遵循雙/超交換機制轉移電子。此外,為了評價Ni3+氧化有機物的實際應用潛力,進一步估算了Ni3+的氧化能力。將NiOOH陽極移至含有有機物的電解液中,Ni3+與有機物發生自發的化學反應,可將NiOOH陽極還原為Ni(OH)2。因此,可以把這個過程分為兩個步驟:一個電化學步驟,在電解液中陰極產生H2和氧化陽極形成NiOOH;隨后一個自發的化學步驟,通過氧化有機物把陽極還原到初始狀態。空間分離的兩步法可以在不同的反應室實現制氫和有機物的氧化,從而有利于生產高純度的產品。Nonredox trivalent nickel catalyzing nucleophilic electrooxidation of organics. Nature Communications, 2023. DOI: 10.1038/s41467-023-43649-6 3. JACS: STM/AFM結合理論計算,證實界面水促進Cu(111)上甲酸去質子化甲酸(FA,HCOOH)是最簡單的羧酸,因其具有較高的容量(53.4 g H2/L)和室溫下的液體性質,在儲氫領域受到越來越多的關注。通常FA的分解包括兩個平行的途徑,即脫氫(產生CO2和H2)和脫水(產生CO和H2O)。值得注意的是,FA在氣相中主要發生脫水反應,但在水相中更易發生脫氫反應,水的存在降低了脫氫反應的反應勢壘。目前大多數的研究都是針對少數FA和水分子進行理論計算。為了全面理解反應機理,必須考慮分子間氫鍵相互作用和分子自組裝以及固體表面的質子轉移過程。由于FA和水分子都是強氫鍵的供體和受體,它們氫鍵相互作用的復雜性可以顯著地決定它們的催化反應。但是,到目前為止,水如何參與催化劑表面上FA的分解仍然不清楚,因此需要在原子尺度上對反應過程進行高分辨率表征。基于此,北京師范大學郭靜、北京理工大學曹端云和南方科技大學郭慶等系統地研究了水對FA在Cu(111)上的表面化學和反應的影響,并證明需要相對較大的水/FA比率來實現FA的完全去質子化和高效的H2生產。具體而言,研究人員使用STM/AFM結合密度泛函理論(DFT)計算,將界面水對Cu(111)上FA去質子化的促進作用可視化,并在原子水平上洞察了反應機理。結果表明,首先,FA與水在Cu(111)表面共吸附時發生解離,生成H+離子和HCOO?離子;同時,大多數水合質子和HCOO?在Cu(111)上表現出相分離行為,其中Eigen和Zundel陽離子組裝成單層六方氫鍵(H-鍵)網絡,并且雙齒HCOO?離子被水溶解并聚集成一維鏈或二維氫鍵網絡。這種相分離行為對于阻止質子從H+離子向HCOO?的轉移和FA的分解是必不可少的。密度泛函理論(DFT)計算表明,水作為酸的強受體(堿),導致了H+離子和HCOO?離子的產生,降低了FA在Cu(111)上的去質子化能壘,其中水通過Grotthuss質子轉移機制催化FA的分解。綜合以上結果,發現需要大量過量的水來促進FA的完全去質子化,其中額外的水作為溶劑,導致形成水-甲酸鹽復合物和離子-水覆蓋物,這也可以解釋為什么需要過量的水來促進FA的完全分解。此外,由于與Cu基底的相互作用增強,H+離子和雙齒HCOO?離子的單獨溶劑化是能量上的優先選擇。此外,程序升溫脫附實驗顯示,水和FA共吸附在Cu(111)表面時,H2脫附峰強度顯著增強,FA脫附降低,進一步表明水對FA脫質子化的促進作用。Visualizing the promoting role of interfacial water in the deprotonation of formic acid on Cu(111). Journal of the American Chemical Society, 2023. DOI: 10.1021/jacs.3c07726 4. JACS: 原子催化劑構型的可逆調整,實現高效氧電還原氧還原反應(ORR)是燃料電池、金屬-空氣電池等可再生能源裝置中必不可少的組成部分。為了推進這些裝置的實際應用,設計一種能夠有效改善ORR緩慢動力學的電催化劑至關重要。具有M-N-C結構的單原子催化劑作為氧還原反應(ORR)的高效電催化劑引起了人們的廣泛關注。值得注意的是,盡管具有相似的M-N-C結構,但是由不同載體(ZIF和石墨相氮g-C3N4)負載的SAC不一定具有相似的催化性能,也就是載體可能導致特征性的金屬載體相互作用(MSI),這可能進一步誘導不同的M-N配位構型,并引起不同的催化行為。因此,在設計SAC時必須考慮這種配置依賴性。然而,在ORR過程中,由衍生框架誘導的SACs的動態M-N構型的作用仍然知之甚少。基于此,臺灣大學陳浩銘和Jiali Wang等制備了一系列固定在g-C3N4和ZIF載體上的具有可控Cu-N構型的Cu SAC作為模型催化劑,來了解動態結構演變和MSIs對ORR催化行為的影響。研究結果表明,g-C3N4上的Cu SAC表現出對稱的Cu-N配位構型,在操作條件下具有可逆的自適應性質,導致其優異的ORR催化活性;相反,ZIF衍生的Cu SAC上的Cu-N構型由于其結構的不對稱性,在ORR過程中發生了不可逆的結構變化,其中拉長的Cu-N對在ORR過程中不穩定且會發生斷裂(該過程與ORR反應競爭),導致ORR的高過電位。除了配位情況,另一個影響ORR活性增強的因素是相互連接的碳骨架,它可以顯著影響電荷轉移過程。與具有相同熱解溫度的裸基底相比,將Cu金屬摻入g-C3N4后,C K邊XAS的光譜特征保持不變;然而,當Cu固定在ZIF骨架上時,局部碳骨架發生重構,導致C=O鍵的富集。值得注意的是,與1s到π*躍遷相關的285.5 eV峰的強度在ZIF衍生的Cu SACs中顯示出顯著的減弱,這種降低可能會降低電荷轉移效率,從而影響ORR性能。總的來說,上述發現為Cu-N構型的動態變化與ORR動力學之間的聯系提供了新見解,并證明了SAC中Cu-N構型的動態適應對于電催化反應的重要性。Reversibly adapting configuration in atomic catalysts enables efficient oxygen electroreduction. Journal of the American Chemical Society, 2023. DOI: 10.1021/jacs.3c10707 5. JACS: 調節催化劑表面Ni3+和OV含量,定向電合成己二酸和環己酮二元羧酸和環酮類化合物,如己二酸(AA)和環己酮(CHN),是化學工業中必不可少的化合物。以水為氧源在溫和條件下將木質素衍生的環己醇(CHA)轉化為CHN和AA被認為是最具競爭力的策略。其中,Ni基催化劑由于其高活性、豐富性和穩定性,被認為是電氧化研究中最有效的電極材料之一。最近,據報道,用十二烷基磺酸鈉(Ni(OH)2-SDS)修飾或摻雜Cu的Ni(OH)2催化劑(Cu-Ni(OH)2)在恒電位(≥1.5 VRHE)下表現出超過80.0%的AA收率。為了達到更高的AA收率(>90%)以滿足當前工業生產的要求,通過研究從CHA到AA的整個反應中涉及的每個步驟(即CHA脫氫和CHN氧化)的機理并同時探索它們的匹配過程來開發更先進的電催化劑至關重要。此外,對CHA脫氫反應的深入了解,有助于控制CHA的電化學反應程度,從而獲得較高的CHN收率,這對于工業上合成己內酰胺(尼龍-6的反應物)、硝化纖維素和涂料等精細化學品和溶劑具有重要意義。近日,復旦大學唐頤、徐昕和暨南大學高慶生等通過金屬基體組分的自溶解、界面生長和電化學活化制備了一系列整體式msig/ea-NiOOH-Ni(OH)2/NF (msig/ea)。光譜表征和理論計算表明,催化劑表面O2?位點可以顯著加速CHA中C-H和O-H鍵的脫氫,而在NiOOH上的OV位點上產生的*OOH物種可以有效提高CHN氧化的活性。通過控制電化學氧化還原活化過程,可以精細地調節表面Ni3+和OV的含量。在優化的活性中心和反應條件下,單步反應(CHA脫氫和CHN氧化)的CHN收率和AA收率分別達到96.5%和93.6%。考慮到最佳電催化劑上CHA轉化為CHN和CHN轉化為AA的選擇性/收率極高,研究人員將CHA脫氫和CHN氧化這兩個體系進行了耦合(以CHA為底物)。結果表明,CHA首先在與Ni3+結合的O2?上脫氫最轉化為CHN,然后形成的CHN在*OOH上氧化并最終獲得了高達92.2%的AA收率,超過了以往所有相關的研究。綜上,這項工作深入研究了分步電化學反應的結構-功能關系,為精確設計更先進的工業催化劑以定向電合成高純度、高附加值的化學品提供了參考。Directional electrosynthesis of adipic acid and cyclohexanone by controlling the active sites on NiOOH. Journal of the American Chemical Society, 2023. DOI: 10.1021/jacs.3c05898 6. JACS: 揭示擴展的不對稱四配位水網絡在HER反應中的主導作用析氫反應(HER)作為一種典型的電化學反應,是理解以水為介質或反應物的各種電化學反應的理論基礎的良好模型,對于將液態水轉化為高價值氫燃料具有重要意義。因此,準確描述電極/溶液界面處的水取向、氫鍵環境和結構轉化對于揭示控制源自水解離和溶劑環境的HER活性的關鍵因素至關重要。最近,氫鍵網絡的連接性和無序性被相繼確定為HER的主導因素。因此,揭示界面水的功能需要精確識別界面水的精細結構和氫鍵網絡的動態變化以及分子水平上雙電層(EDL)中復雜的相互作用,但這仍然是一個挑戰。最近,清華大學李景虹和中國科學院長春應化所姜秀娥等結合原位SEIRA光譜電化學、1H核磁共振波譜和MD模擬,揭示了水結構與HER活性之間的關系。研究發現,水分子從孤立到水團簇甚至更大的氫鍵網絡的演化將導致HER活性的逐漸增強。在高負極化電位下,水分子的O-H鍵會斷裂,形成H*中間體和OH?離子;由此產生的電荷被直接與Au膜相互作用的相對無序的非對稱四配位水網絡傳輸離開界面,而具有強氫鍵的對稱四配位水網絡由于其剛性和遠離界面的特點,可能不利于HER過程。這成功區分了與電極相互作用的水和外層水,闡明了不同結構的水在電化學反應中的重要作用,對于理解以水為反應物或介質的電化學反應的基本步驟具有重要意義。此外,通過添加親水性和疏水性陽離子,發現親水性Li+離子在短程范圍內會促進水的解離,但在長程范圍內會破壞水網絡的連通性;而疏水性三丁基溴化銨陽離子則極大地促進了界面處形成擴展的不對稱四配位網絡,有利于OH?離子通過EDL的Volmer步驟傳輸,從而增強了HER。因此,只有離子-水局域相互作用和氫鍵網絡連接性的協同作用才能主導最佳的水解離和電荷傳輸,從而獲得最高的HER活性。綜上,該項工作清楚地揭示了依賴于電位的不對稱四配位水網絡的連接性受親水和疏水陽離子的調控,并與HER活性正相關,為揭示水在催化、能源和表面科學中的功能提供了參考。Uncovering the dominant eole of an extended asymmetric four-coordinated water network in the hydrogen evolution reaction. Journal of the American Chemical Society, 2023. DOI: 10.1021/jacs.3c08333 7. ACS Catal.: Cu基電催化劑表面羥基化,促進電化學還原CO2電化學CO2還原反應(CO2RR)為CO2直接轉化為高附加值化學品和燃料提供了一條可持續的途徑。在眾多CO2RR產物中,多碳(C2+)產品由于其與C1產品相比具有更高的能量密度和市場價值而受到廣泛關注。由于Cu表面上最佳的*CO結合能,其可作為具有最佳C2+產物選擇性的CO2RR催化劑。迄今為止,在高性能銅基電催化劑的結構設計和精確制備方面取得了重大進展。并且,許多工作已經開始著手研究與電極界面微環境調控相關的一些關鍵因素和策略,如催化劑表面的分子工程、電解質設計和電解池構型等,這促使人們在設計和制備催化劑以引導對目標產物的選擇性時應始終考慮催化劑周圍的反應微環境。目前,研究較多的策略之一是利用濃KOH將CO2地轉化為C2+產物,但陰極上碳酸鹽的生成消耗了大部分輸入的CO2。因此,尋求一種新的策略來改善電極局部環境,從而消除或降低CO2RR選擇性對本體OH?濃度的依賴至關重要。基于此,華東理工大學李春忠、李會會和日本東北大學李昊等通過羥基功能化的表面策略(即在Cu2O催化劑上覆蓋上富含羥基的分子)來實現分子表面修飾以增強C2+產物的形成。電化學實驗和原位表征證實了在CO2RR反應過程中,催化劑表面附近穩定存在的羥基物種能夠有效地將吸附的*CO轉化為C2+產物。在流動池中,最優的0D-Cu-800催化劑上C2+產物的法拉第效率為81.5%,部分流電流密度為285 mA cm?2,以及陰極能量效率為43.1%;利用陽離子交換膜電極組件裝置,研究人員證明了在平均電流密度為151 mA cm?2的條件下,0D-Cu-800上能夠連續穩定生產C2H4超過100小時。理論計算表明,富含羥基的分子如葡萄糖酸可以導致Cu位點的電子損失,這有助于改善CO與Cu之間的電子轉移,進而促進*CO的吸附和C-C耦合,提高了電化學CO2RR對C2+產物的選擇性。總的來說,該項工作證明了表面修飾對于設計穩定的反應微環境的重要性,這為未來設計高效的電催化劑以提高CO2RR或其他電化學反應活性提供了指導。Boosting electrochemical CO2 reduction via surface hydroxylation over Cu-based electrocatalysts. ACS Catalysis, 2023. DOI: 10.1021/acscatal.3c02454 8. Adv. Sci.: F遷移耦合雙金屬中心,促進材料表面重構以提升OER活性電化學水分解是一種生產高純度氫氣的有效途徑。然而,在陽極半反應中,由于多步質子/電子耦合,析氧反應(OER)比陰極析氫反應(HER)需要更大的過電位,這阻礙了能量的有效轉化。其中,RuO2和IrO2等貴金屬催化劑能夠顯著降低OER的過電位,但其昂貴的價格和稀缺的儲量嚴重限制了它們的大規模工業應用。在眾多貴金屬催化劑替代品中,過渡金屬基電催化劑由于其價廉、儲量豐富等優點而得到了廣泛的研究。其中,過渡金屬氟化物中的F具有最大的電負性,可以將金屬中心的價態推向最高,這有助于輔助催化劑表面重構、形成非晶結構等。因此,氟化物可能是OER的有效催化劑,但目前文獻中很少有關其的報道。近日,武漢大學趙蘋蘋和程功臻等設計并制備了一種結晶性良好的NiCo雙金屬氟化物(Ni0.42Co0.58F2-G),并研究了其在電化學過程中的重構現象。具體而言,研究人員首先采用固液交換法合成了具有較大比表面積的NiCo基納米陣列A-Ni-MeIM-Co-0.01前體,再通過一步氣相氟化法將其轉化為具有較高電化學活性表面積的NiCo雙金屬氟化物Ni0.42Co0.58F2-G。與傳統的液相合成方法相比,氣相氟化法生產的氟化物具有更高的結晶度;同時,雙金屬中心電子結構的調控在電化學過程中促進了高效的表面重構以及重構過程中F的遷移導致更多電化學活性位點的暴露。這不僅使Ni0.42Co0.58F2-G具有優異的電化學性能,而且保證了其電化學穩定性。電化學性能測試結果顯示,所制備的Ni0.42Co0.58F2-G催化劑在10 mA cm?2電流密度下的OER過電位僅為313 mV,Tafel斜率為42.5 mV dec?1;并且,該催化劑在10000次CV循環后活性仍保持穩定,表明其具有優異的穩定性。此外,理論計算表明,雙金屬中心的構建可以誘導電荷重新分布并產生更高價金屬位點,從而優化關鍵反應中間體的吸附,降低反應能壘,顯著提高OER活性。綜上,該項工作詳細研究了F和雙金屬中心在提升催化劑OER活性中的作用,這對于設計和制備高效的電化學表面重構電催化劑具有一定的指導意義。Surface reconstruction facilitated by fluorine migration and bimetallic center in NiCo bimetallic fluoride toward oxygen evolution reaction. Advanced Science, 2023. DOI: 10.1002/advs.202306758【做計算 找華算】華算科技專注DFT代算服務、正版商業軟件版權、全職海歸計算團隊,10000+成功案例!客戶成果發表在Nature、Nature Catalysis、JACS、Angew.、AM、AEM、AFM等頂刊,好評如潮,專業靠譜!添加下方微信好友,立即咨詢: